
La Lettre de la NSFA n° 43, avril 2009
A. Kontush, M.-J. Chapman, INSERM Unité 939 « Dyslipidémies, Inflammation et Athérosclérose dans les Maladies Métaboliques », Hôpital de la Pitié, Paris
Résumé
Les 10 dernières années ont vu une explosion du nombre d’études portant sur le rôle des lipoprotéines dans les fonctions cérébrales. Il a été de plus en plus reconnu que des anomalies du métabolisme cérébral des lipides étaient étroitement liées à la pathogenèse d’atteintes neurodégénératives majeures, telles que la maladie d’Alzheimer (MA) et la démence vasculaire (DV). Cet article discute les données actuellement disponibles, en particulier à la lumière d’une étude récemment publiée (Whitehall II study), qui suggère une association entre des taux plasmatiques bas de HDL-cholestérol et un déficit de la mémoire verbale à court terme chez des adultes d’âge moyen.
Les 10 dernières années ont vu une explosion du nombre d’études portant sur le rôle des lipoprotéines dans les fonctions cérébrales. Les neurones nécessitent un apport continu de lipides pour la synthèse membranaire et la production d’acétylcholine. Le cerveau est donc le siège d’un turn-over élevé des lipides – bien que le système nerveux central (SNC) ne représente que 2,1 % du poids corporel, il contient 23 % du cholestérol de l’ensemble du corps humain [1]. Le métabolisme lipidique dans le cerveau est étroitement contrôlé localement, car les lipoprotéines plasmatiques sont isolées du cerveau par la barrière hématoencéphalique (BHE). Bien que les cellules neuronales soient capables d’une synthèse de novo d’un large spectre d’espèces moléculaires de lipides, elles dépendent fortement de sources exogènes et lient et internalisent facilement les lipoprotéines des liquides extracellulaires [2]. De même, les neurones ont besoin d’évacuer les lipides en excédant ; le transport des lipides par les lipoprotéines est donc bidirectionnel et inclut un efflux cellulaire de cholestérol [3]. Le liquide céphalo-rachidien (LCR) humain contient essentiellement des lipoprotéines sphériques, d’approximativement 10-12 nm de diamètre, d’une densité de l’ordre de 1,063-1,25 g/ml, ressemblant ainsi aux particules HDL (High density lipoproteins) du plasma humain [3] [4]. Les concentrations des lipides dans le LCR sont cependant beaucoup plus faibles (environ 300-400 fois moindre pour le cholestérol total et les phospholipides) par comparaison au compartiment plasmatique [4]. Les apolipoprotéines (apo) E et A-I sont les principales apolipoprotéines dans le LCR humain (fourchette de concentration habituelle : 0,1-0,4 mg/dl [3, 4]), avec des apoA-II, A-IV, J, D, C-II, C-III, C-IV et H également présentes [3, 4]. Il est important de souligner que les lipoprotéines du LCR transportent la substance amyloïde- β (Aβ), un peptide de 39 à 43 acides aminés, produit dans les cellules neuronales, et qui est le composant principal des plaques amyloïdes séniles [5].
Le métabolisme des lipoprotéines du LCR demeure mal connu, mais semble être différent de celui des lipoprotéines plasmatiques. Les particules HDL enrichies en apoE sont synthétisées dans le SNC et sécrétées par les astrocytes sous forme de complexes discoïdaux enrichis en cholestérol libre [6]. Leur forte teneur en apoE amène ces lipoprotéines à se lier aux récepteurs cellulaires aux apoE, auxquels elles se lient avec une forte affinité, en particulier la LDL receptor-related protein, qui est exprimé en abondance à la surface des neurones [2]. En revanche, les lipoprotéines du LCR enrichies en apoA-I sont vraisemblablement dérivées du HDL plasmatique qui pénètre dans le SNC en traversant la BHE [7], car l’apoA-I n’est pas synthétisée dans le SNC [2]. Tout comme les lipoprotéines plasmatiques, les lipoprotéines du LCR peuvent être remodelées par différentes protéines, la lecithin-cholesterol acyltransferase (LCAT) et la phospholipid transfer protein (PLTP) ; cependant, la cholesteryl ester transfer protein (CETP) est absente du LCR [3]. Ces données démontrent clairement que les particules HDL du cerveau constituent un élément clé de l’homéostasie du cholestérol dans le SNC.
Durant ces 10 dernières années, il a été de plus en plus reconnu que les anomalies du métabolisme cérébral des lipides étaient étroitement liées à la pathogenèse d’atteintes neurodégénératives majeures, telles que la maladie d’Alzheimer (MA) et la démence vasculaire (DV). Avec des fréquences représentant respectivement 70 % et 15 % de toutes les démences, la MA et la DV sont les formes les plus communes de démences et sont habituellement précédées par un déclin cognitif progressif, incluant une diminution de la mémoire [8]. Tant la MA que l’athérosclérose, se développent parallèlement au cours du processus de vieillissement, et de ce fait, ont en commun de nombreux facteurs de risque, incluant l’existence d’un diabète de type 2 et des taux élevés de cholestérol total et de lipoprotéine (a) – Lp(a) – chez des sujets d’âge moyen, ce qui suggère des voies physiopathologiques communes [9]. Bien entendu, la progression à la fois de la MA et de l’athérosclérose dans les modèles animaux est totalement dépendante de l’expression d’apolipoprotéines spécifiques, telle que l’apoE ; de plus, ces deux maladies impliquent une inflammation locale chronique, soit dans le cerveau, soit dans la paroi artérielle, qui est un facteur pathologique caractéristique et crucial.
Il est parfaitement démontré qu’un taux bas de HDL-cholestérol (HDL-C) est un facteur de risque majeur de maladies cardiovasculaires. En conséquence, les particules HDL font actuellement l’objet de nombreux travaux de recherche, car elles représentent une cible thérapeutique prometteuse pour la prévention, ou la réduction, des maladies cardiovasculaires [10]. Cependant, la question qui intrigue les spécialistes des lipides est : « La fonction protectrice des particules HDL demeure t-elle vraie dans la MA, la DV et les autres types de démences ? ». La récente étude de Singh-Manoux et al. (Whitehall II study) [11] suggère une association entre des taux plasmatiques bas de HDL-C et un déficit de la mémoire verbale à court terme chez des adultes d’âge moyen. De plus, la diminution du taux de HDL-C, durant un suivi de 5 ans, est associée à un déclin de la mémoire. Ces associations persistent après ajustements sur le niveau d’éducation, le niveau socioprofessionnel, les maladies associées et les traitements suivis. Il est intéressant de constater que les associations entre des taux plasmatiques bas de HDL-C et les déficits mnésiques sont indépendants de la présence de l’allèle apoE4, un important facteur de risque de la MA. Par contraste, il n’apparaît pas d’associations significatives entre les concentrations circulantes de cholestérol total, de triglycérides et le déclin ou déficit mnésique. Ces résultats, qui sont sans aucun doute d’un très grand intérêt, donnent-ils une place indiscutable au HDL-C dans nos mémoires ?
Bien que les études observationnelles conduites antérieurement aient abouti à des résultats discordants en ce qui concerne les liens entre les taux de HDL-C et la fonction cognitive [revue in [12], [13], des concentrations basses de HDL-C ont été fréquemment rapportées comme étant associées à la démence. La majorité des études conduites jusqu’à maintenant ont été transversales, l’étude de Singh-Manoux et al. [11] est donc clairement différente car elle consiste en l’évaluation de l’association d’une étude transversale et d’une étude prospective. Par ailleurs, les taux élevés plasmatiques de HDL-C, potentiellement médiés par une activité CETP faible, sont également associés à la longévité, à une cognition améliorée et à la survie indemne de démence [14]. À l’inverse, les polymorphismes de la CETP conduisant à un HDL-C bas sont fortement présents dans la MA [15]. Cependant, aucune de ces études n’infère un lien de causalité car, ainsi que Singh-Manoux et al. l’indiquent [11], les taux de lipides plasmatiques peuvent se modifier considérablement au cours du développement et de la progression de la démence, ce qui implique que le moment précis des mesures est un point critique.
Toutefois, la question clé qui n’est pas étudiée par les auteurs est : quels sont les mécanismes potentiels à l’origine de ces effets ? Singh-Manoux et al. [11] mentionnent différentes propriétés neuroprotectrices du HDL, incluant une maturation accélérée des synapses, le maintien de la plasticité synaptique, un métabolisme amélioré de Aβ et une augmentation du volume de l’hippocampe, et on peut ajouter à cette longue liste, des propriétés antiinflammatoires et antioxydantes – ceci uniquement pour illustrer toute la complexité et la variabilité des mécanismes biochimiques qui, potentiellement, relient HDL et MA.
La complexité de la relation mécanistique entre les particules HDL et fonction cérébrale apparaît immédiatement lorsque l’on considère le métabolisme de Aβ, la voie majeure impliquée dans la pathogenèse de la MA. Les particules HDL du cerveau peuvent en effet exercer plusieurs effets neuroprotecteurs en agissant uniquement via cette voie métabolique (figure 1).
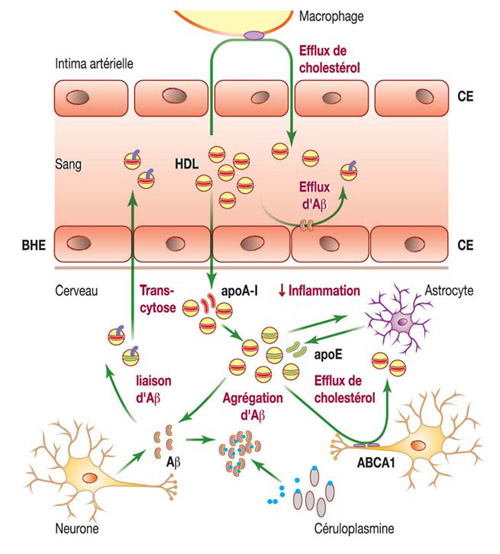
Figure 1 : Mécanismes potentiels pouvant expliquer le rôle neuroprotecteur du HDL.
Aβ : peptide amyloïde-β ; ABCA1 : ATP-binding cassette transporter A1 ; BHE : barrière hématoencéphalique ; CE : cellules endothéliales.
- Comme la production neuronale de Aβ est fréquemment parallèle au contenu membranaire en cholestérol, les particules HDL peuvent réduire la production d’Aβ en diminuant le cholestérol intracellulaire par l’activation du transport inverse du cholestérol médié par les transporteurs ABC (ATP-binding cassette transporter) [16].
- Les particules HDL peuvent directement se lier à l’Aβ en excès, et de ce fait, inhiber son oligomérisation [17], cette dernière représentant une étape capitale dans la transformation du monomère Aβ non toxique en une forme agrégée neurotoxique qui peut être en cause dans l’altération de la mémoire [18]. Comme les particules HDL transportent Aβ à la fois dans le LCR et dans le plasma [5], l’élimination du peptide se trouvant en excès dans le cerveau peut s’ensuivre. De plus, les particules HDL pourraient également capter l’Aβ qui s’accumule dans les parois vasculaires au cours de la DV [19] ; par analogie avec le transport inverse du cholestérol, un tel processus peut être dénommé « transport inverse d’amyloïde-β ».
- Le stress oxydant induit une production d’Aβ, une réponse potentiellement protectrice ; en effet, l’Aβ monomérique est un chélateur particulièrement puissant des ions des métaux de transition qui sont pro-oxydants en leur forme libre [20] ; en retour, les particules HDL peuvent diminuer le stress oxydant [21] et ainsi, diminuer indirectement la production d’Aβ.
Les particules HDL peuvent agir sur les astrocytes pour atténuer une réaction inflammatoire locale.
Dans tous ces scénarios, il demeure peu clair comment des taux bas de HDL-C mesurés dans le plasma peuvent être traduits en un défaut de fonctionnalité du HDL dans le cerveau. L’apoA-I, composant majeur du HDL plasmatique, impliquée dans l’efflux de cholestérol cellulaire et dans d’autres activités biologiques du HDL, pourrait représenter un lien potentiel [22]. Les taux plasmatiques de HDL-C et d’apoA-I sont fortement corrélés dans la population générale [23] ; il est donc possible que les taux d’apoA-I diminuent en parallèle à ceux de HDL-C chez les sujets atteints d’un déficit mnésique. L’élimination du cholestérol des cellules neuronales, parallèle à la réduction de la neuro-inflammation, toutes deux médiées par l’apoA-I capable de traverser la BHE, pourrait donc, d’un point de vue mécanistique, expliquer la relation entre les taux plasmatiques faibles de HDL-C et les déficits mnésiques observés [11].
Enfin, l’inhibition de l’athérosclérose des gros vaisseaux par les particules HDL est une autre possibilité intéressante à considérer puisque les pathologies vasculaires peuvent jouer un rôle direct dans l’initiation des déficits neurologiques de la MA et de la DV.
Singh-Manoux et al. [11] soulignent certaines limitations de leur étude, incluant son caractère observationnel, des ajustements potentiellement incomplets pour des facteurs confondants (tels que la consommation tabagique, la consommation d’alcool habituelle et l’exercice physique, tous facteurs influençant fortement les taux de HDL-C), le niveau socioéconomique élevé des participants et leur survie potentielle, ainsi que des biais potentiels compte tenu de la survie des sujets et de la durée du suivi. Cette liste devrait également être étendue à l’absence de distinction entre l’état à jeun ou non des sujets, tout comme à l’absence de distinction entre hommes et femmes.
En effet, il est évident que les taux de HDL-C dépendent étroitement du sexe et peuvent être modifiés en phase postprandiale [24]. La faible proportion de femmes dans la population étudiée (< 30 %) pose la question de la généralisation de ces résultats à la population féminine. De plus, une présentation plus conventionnelle, des taux plasmatiques de lipides, sous forme de variables continues, plutôt qu’une catégorisation, aurait pu apporter des informations plus détaillées sur leurs relations avec les déficits mémoriels. Un autre point observé dans cette étude, la diminution du risque cardiovasculaire concomitant à l’augmentation des taux de HDL-C et à la diminution du cholestérol total avec l’âge [11], n’est pas un constat classique, et est plus probablement lié à l’utilisation élevée (x 3,7) de médicaments hypolipémiants ; une telle limitation devrait, de plus, avoir pour conséquence de réduire le nombre de sujets présentant une diminution de leur concentration plasmatique de HDL-C durant le suivi, et de ce fait, se traduire par une réduction de la puissance statistique.
Enfin, l’absence de données sur l’incidence du diabète et de l’obésité dans la population de Whitehall 2 [11] est regrettable. Le diabète de type 2 partage des caractéristiques métaboliques communes avec la MA, et notre connaissance de nombre de voies métaboliques communes à ces deux pathologies majeures évolue progressivement – par exemple, des taux élevés d’insulinémie résultent en une production élevée de peptides Aβ [25]. Des taux de HDL-C inférieurs à la normale sont fréquemment constatés dans le diabète de type 2 [24], donc l’augmentation de la prévalence de cette maladie chez des sujets à HDL-C bas, pourrait être la cause sous-jacente de l’association avec le déclin de la mémoire observé par Singh-Manoux et al. [11]. En tout état de cause, le lien entre diabète de type 2, HDL-C bas et déclin mnésique justifie d’autres études.
Il est tentant de spéculer que des taux élevés de HDL-C, le « bon cholestérol », pourraient protéger notre mémoire, et les auteurs n’échappent pas à cette tentation. Cependant, les résultats décevants des larges études d’interventions conduites avec des supplémentations par des antioxydants d’origine alimentaire [26], suggèrent que nous devons demeurer extrêmement prudents avant de proposer des interventions thérapeutiques sur la base d’études observationnelles qui n’impliquent pas un lien de causalité. Ceci est particulièrement vrai dans le cas d’une étude qui a de nombreuses et importantes limitations, telle que celle de Singh-Manoux et al. [11].
Par exemple, une étude attentive de leurs données révèle, qu’alors que la chute du taux d’HDL-C au cours de l’étude était associée à une détérioration de la mémoire – par comparaison à leur stabilité, une augmentation des concentrations de HDL-C n’était pas associée à une amélioration de la mémoire [11]. Au minimum, de telles données observationnelles devraient être documentées par l’utilisation d’approches neuroanatomiques et neurofonctionnelles directes, plutôt que par de simples tests de mémoires.
Conclusion
Si le HDL-C demeure une cible potentiellement prometteuse pour la prévention de la démence et de la perte de mémoire, celle-ci demeure encore lointaine. Néanmoins, ces études justifient que davantage d’efforts de recherche soient portés sur les relations liant HDL et fonction cérébrale.
Les points essentiels
- Le métabolisme des lipoprotéines du liquide céphalo-rachidien demeure mal connu, mais semble être différent de celui des lipoprotéines plasmatiques.
- Les données actuelles démontrent clairement que les particules HDL du cerveau constituent un élément clé de l’homéostasie des lipides dans le système nerveux central.
- La récente étude de Singh-Manoux et al. (Whitehall II study) suggère une association entre des taux plasmatiques bas de HDL-cholestérol (HDL-C) et un déficit de la mémoire verbale à court terme chez des adultes d’âge moyen.
- Il est donc tentant de spéculer que des taux élevés de HDL-C, le « bon cholestérol », pourraient protéger notre mémoire. Cependant, nous devons demeurer extrêmement prudents avant de proposer des interventions thérapeutiques sur la base d’études observationnelles qui n’impliquent pas un lien de causalité.
- Si le HDL-C demeure une cible potentiellement prometteuse pour la prévention de la démence et de la perte de mémoire, celle-ci demeure encore lointaine.
[1] Dietschy JM, Turley SD. Thematic review series : Brain lipids. Cholesterol metabolism in the central nervous system during early development and in the mature animal. J Lipid Res 2004 ;45:1375-97.
[2] Beffert U, DanikM, Krzywkowski P, et al. The Danik M, neurobiology of apolipoproteins and their receptors in the CNS and Alzheimer’s disease. Brain Res Brain Res Rev 1998 ;27:119-42.
[3] Demeester N, Castro G, Desrumaux C, et al. Characterization and functional studies of lipoproteins, lipid transfer proteins, and lecithin : cholesterol acyltransferase in CSF of normal individuals and patients with Alzheimer’s disease. J Lipid Res 2000 ;41:963-74.
[4] Koch S, Donarski N, Goetze K, et al. Characterization of four lipoprotein classes in human cerebrospinal fluid. J Lipid Res 2001 ;42:1143-51.
[5] Kontush A. Apolipoprotein Abeta : black sheep in a good family. Brain Pathol 2004 ;14:433-47.
[6] Ladu MJ, Reardon C, Van Eldik L, et al. Lipoproteins in the central nervous system. Ann N Y Acad Sci 2000 ;903:167-75.
[7] Balazs Z, Panzenboeck U, Hammer A, et al. Uptake and transport of high-density lipoprotein (HDL) and HDL-associated alpha-tocopherol by an in vitro blood-brain barrier model. J Neurochem 2004 ;89:939-50.
[8] Whitehouse PJ, Sciulli CG, Mason RM. Dementia drug development : use of information systems to harmonize global drug development. Psychopharmacol Bull 1997 ;33:129-33.
[9] Martins IJ, Hone E, Foster JK, et al. Apolipoprotein E, cholesterol metabolism, diabetes, and the convergence of risk factors for Alzheimer’s disease and cardiovascular disease. Mol Psychiatry 2006 ;11:721-36.
[10] Kontush A, Guerin M, Chapman MJ. Spotlight on HDL-raising therapies : insights from the torcetrapib trials. Nat Clin Pract Cardiovasc Med 2008 ;5:329-36.
[11] Singh-Manoux A, Gimeno D, Kivimaki M, et al. Low HDL cholesterol is a risk factor for deficit and decline in memory in midlife : the Whitehall II study. Arterioscler Thromb Vasc Biol 2008 ;28:1556-62.
[12] Michikawa M. Cholesterol paradox : is high total or low HDL cholesterol level a risk for Alzheimer’s disease ? J Neurosci Res 2003 ;72:141-6.
[13] Panza F, D’Introno A, Colacicco AM, et al. Lipid metabolism in cognitive decline and dementia. Brain Res Rev 2006 ;51:275-92.
[14] Barzilai N, Atzmon G, Derby CA, et al. A genotype of exceptional longevity is associated with preservation of cognitive function. Neurology 2006 ;67:2170-5.
[15] Chen DW, Yang JF, Tang Z, et al. Cholesteryl ester transfer protein polymorphism D442G associated with a potential decreased risk for Alzheimer’s disease as a modifier for APOE epsilon4 in Chinese. Brain Res 2008 ;1187:52-7.
[16] Wahrle SE, Jiang H, Parsadanian M, et al. Overexpression of ABCA1 reduces amyloid deposition in the PDAPP mouse model of Alzheimer disease. J Clin Invest 2008 ;118:671-82.
[17] Olesen OF, Dago L. High density lipoprotein inhibits assembly of amyloid beta-peptides into fibrils. Biochem Biophys Res Commun 2000 ;270:62-6.
[18] Lesné S, Koh MT, Kotilinek L, et al. A specific amyloid-beta protein assembly in the brain impairs memory. Nature 2006 ;440:352-7.
[19] Hardy J, Cullen K. Amyloid at the blood vessel wall. Nat Med 2006 ;12:756-7.
[20] Kontush A. Amyloid-beta : an antioxidant that becomes a pro-oxidant and critically contributes to Alzheimer’s disease. Free Radic Biol Med 2001 ;31:1120-31.
[21] Kontush A, Chantepie S, Chapman MJ. Small, dense HDL particles exert potent protection of atherogenic LDL against oxidative stress. Arterioscler Thromb Vasc Biol 2003 ;23:1881-8.
[22] Scarmeas N. Invited commentary : lipoproteins and dementia – is it the apolipoprotéine A-I ? Am J Epidemiol 2007 ;165:993-7.
[23] Ridker PM, Rifai N, Cook NR, et al. Non-HDL cholesterol, apolipoproteins A-I and B100, standard lipid measures, lipid ratios, and CRP as risk factors for cardiovascular disease in women. JAMA 2005 ;294:326-33.
[24] Lamarche B, Rashid S, Lewis GF. HDL metabolism in hypertriglyceridemic states : an overview. Clin Chim Acta1999 ;286:145-61.
[25] Li L, Holscher C. Common pathological processes in Alzheimer disease and type 2 diabetes : a review. Brain Res Rev 2007 ;56:384-402.
[26] Bjelakovic G, Nikolova D, Gluud LL, et al. Mortality in randomized trials of antioxidant supplements for primary and secondary prevention : systematic review and meta-analysis. JAMA 2007 ;297:842-57.