
Les cardiopathies ischémiques et les accidents vasculaires cérébraux sont parmi les premières causes de mortalité dans le monde (respectivement 7,4 et 5,1 millions de décès en 1998 selon le rapport sur la santé dans le monde de l’OMS de 1999). Leur prévalence devrait croître dans les prochaines années en raison de l’adoption par des populations jusque-là épargnées de modes de vie occidentaux comportant des facteurs de risque inhérents. Ces modifications ont contribué à une pandémie de diabète de type II et de syndrome métabolique, tous deux impliquant des facteurs de risque cardiovasculaire tels que insulino-résistance, obésité viscérale, hypertension et dyslipidémie athérogène. Actuellement, 50 millions d’individus en Amérique du Nord sont atteints de syndrome métabolique [1].
Description anatomopathologique de l’athérosclérose
Les accidents ischémiques aigus (mort subite, infarctus myocardique ou cérébral, angor instable) sont, dans la majorité des cas, la traduction clinique de la maladie athéromateuse, consécutifs à une rupture ou une érosion de la plaque d’athérosclérose avec formation d’un thrombus obstruant la lumière vasculaire [2].
L’athérosclérose n’est pourtant pas une maladie des temps modernes, puisque les plaques d’athérome authentiques sur le plan histologique ont été identifiées au sein des corps momifiés égyptiens [3].
Antonio Scarpa (1752-1832), dans son travail sur l’anévrisme artériel (1804), est le premier à décrire une maladie de la couche interne des grosses artères que l’on nomme aujourd’hui athérosclérose [4] [5]. Pour lui, l’anévrisme de l’aorte résulte « d’une dégénérescence de la couche interne de l’artère, lente, à ulcération pathologique stéatomateuse, fongueuse et squameuse ».
Le terme d’athérome, du grec athara : « bouillie de farine ou de gruau », est proposé pour la première fois par Albrecht von Haller en 1755. En 1833. Lobstein préfère utiliser artériosclérose considérant que les altérations artérielles ne sont pas d’origine inflammatoire et sont dues à une mauvaise nutrition des vaisseaux. Au contraire, Virchow, en 1856, parle d’endartérite déformante pour souligner le caractère inflammatoire de la maladie. Councilman, en 1891, propose artériosclérose nodulaire, repris plus tard par Osler (1897). C’est finalement Marchand, en 1904, qui invente le terme d’athérosclérose qui reflète la dualité lésionnelle, athéromateuse et scléreuse, de la maladie.
La description anatomopathologique moderne de l’athérosclérose retient trois stades évolutifs : strie lipidique, lésion fibro-lipidique et lésion compliquée.
Stary [6] a proposé une classification plus détaillée, reposant sur l’observation d’un grand nombre d’artères d’enfants et d’adultes jeunes, qui divise les événements pathologiques en sept stades de gravité croissante (tableau 12-1). Elle suggère que les lésions évoluent avec l’âge du sujet en passant successivement d’un type lésionnel au type immédiatement supérieur. Un élément clé dans notre compréhension des événements précoces a été apporté en 1989 par les études de Schwenke et Carew [7] [8]. Ils ont mis en évidence une accumulation pré-lésionnelle de lipoprotéines de basse densité riches en cholestérol athérogènes, telles que les LDL, liées aux protéoglycanes de la matrice, au niveau des sites de prédilection de la formation des plaques d’athérome.
Par ailleurs, les progrès accomplis dans la détection et la caractérisation des composants cellulaires de la plaque d’athérome, en particulier grâce à l’immunohistochimie, ont permis de lever un grand nombre d’incertitudes (tableau 12-2) [9]. Les cellules spumeuses de la strie lipidique, caractéristique de la plaque au stade précoce de son développement, sont d’origine essentiellement macrophagique. En fait, c’est la modification de la structure des LDL retenues dans la matrice extracellulaire (par oxydation, par exemple) qui conduit à leur internalisation par les monocytes-macrophages, qui se transforment en cellules spumeuses suite à l’accumulation intracellulaire de gouttelettes d’ester de cholestérol [10]. A une étape plus avancée du processus athéromateux (stade IV/V, tableau 12-2), une chape fibreuse constituée principalement de cellules musculaires lisses (CML) vient entourer la masse lipidique, donnant naissance à la plaque fibro-lipidique. De plus, des lymphocytes T sont également présents en quantité assez abondante, en bordure de la plaque et dans la chape fibreuse [11].
Tableau 12-1 • Classification des lésions de l’athérosclérose, d’après Stary [6].
TYPE LÉSIONEL
I
II
III
IV
V
VI
VII
TERME PROPOSÉ
Macrophages spumeux isolés
Strie lipidique
Préathérome
Athérome
Plaque athéroscléreuse
Plaque athéroscléreuse compliquée
Plaque fibreuse
DESCRIPTION
Macrophages spumeux isolés dans l’intima. Absence de lipides extracellulaires.
Couches de macrophages spumeux. Cellules musculaires lisses dans l’intima chargées de lipides. Fines particules lipidiques extracellulaires disséminées.
Modifications de type II associées à de multiples dépôts lipidiques extracellulaires formant de petits agrégats.
Modifications de type Il associées à de multiples dépôts lipidiques extracellulaires massifs et confluents (noyau lipidique).
Modifications de type IV associées à des dépôts massifs de collagène (chape fibreuse) recouvrant le noyau lipidique (type Va), avec calcifications (type Vb).
Modifications de type V avec rupture de la chape fibreuse (VIa), hémorragie intraplaque (VIb) ou thrombose (VIc).
Épaississement massif de l’intima par sclérose collagène ; lipides intra- et extracellulaires absents ou présents en quantité négligeable.
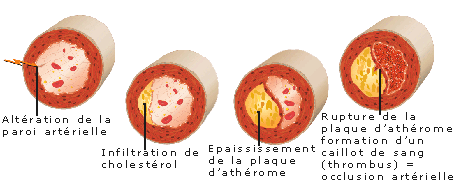
Tableau 12-2. Composition cellulaire de la plaque d’athérosclérose chez l’homme (carotide interne), en pourcentage du nombre total de cellules (d’après Jonasson et coll. [9]).
Macrophages
Cellules musculaires lisses
Lymphocytes T
CHAPE FIBREUSE
24
60
16
NOYAU LIPIDIQUE
60
29
10
Les différentes théories de l’athérogenèse
Le pathologiste Rudolf Virchow (1821-1902) a développé deux théories sur la pathogenèse et l’étiologie de l’athérosclérose [12]. Selon lui, les lipides plasmatiques sont absorbés directement à travers l’endothélium par l’intima et, lorsque les conditions le permettent, certains d’entre eux, en particulier les esters de cholestérol, s’y déposent. C’est l’hypothèse de l’infiltration lipidique comme facteur déclenchant la formation de la plaque d’athérome, confortée au début du siècle par les travaux d’Anitschkow et Chalatow (1913) qui mirent en évidence le rôle du cholestérol dans l’athérosclérose expérimentale chez le lapin [4].
Virchow pensait également qu’un traumatisme endothélial provoqué d’origine mécanique était à l’origine de l’infiltration lipidique. Sur cette deuxième hypothèse, Russell Ross a développé dès 1976 la théorie de la « réponse à l’effraction endothéliale » [13], selon laquelle une lésion de l’endothélium serait le primum movens de l’athérosclérose. Or, cette conception de l’athérosclérose s’est révélée totalement inexacte. Aucun processus de dégénérescence, de rupture ou de desquamation endothéliale pouvant être à l’origine du processus athéroscléreux n’a jamais pu être mis en évidence [14]. L’endothélium conserve son intégrité structurale au cours du développement de l’athérosclérose, mais il présente un état d’activation inflammatoire favorisant le recrutement des monocytes et des lymphocytes circulants. De plus, les plaquettes n’interviennent pas dans la genèse de la plaque. Elles participent, en partie, à la formation du thrombus fibrino-plaquettaire après rupture ou érosion de la plaque, entraînant la survenue d’un syndrome coronarien aigu. Enfin, la prolifération des CML n’est en rien délétère elle constitue une étape essentielle de la réparation tissulaire [15]. Les CML assurent la stabilité de la lésion : les plaques riches en CML et en collagène, et pauvres en macrophages sont cliniquement silencieuses [16].
Les études expérimentales les plus récentes, associées aux observations anatomopathologiques faites sur des plaques humaines, permettent d’affirmer aujourd’hui que l’athérosclérose est une maladie inflammatoire chronique des grosses artères à localisation intimale [17] [18], et que l’agent d’agression entraînant la réaction inflammatoire est très probablement le cholestérol-LDL modifié, notamment par oxydation [10].
Rôle des LDL et des lipoprotéines athérogènes dans l’athérosclérose
PÉNÉTRATION ET RÉTENTION DES LDL DANS L’INTIMA
L’importance du cholestérol et plus particulièrement des LDL dans l’athérogenèse n’est plus contestée, depuis que les essais cliniques de prévention primaire et secondaire chez les sujets hypercholestérolémiques ont démontré qu’il était possible de réduire la fréquence des cardiopathies ischémiques en diminuant le cholestérol-LDL à l’aide de statines [19].
Avec les lipoprotéines de très basse densité, les remnants de VLDL (VLDLR), les lipoprotéines de densité intermédiaire et la lipoprotéine Lp(a), les LDL font partie des lipoprotéines athérogènes dont les taux sont étroitement associés au risque cardiovasculaire et à une athérosclérose précoce [10] [20] [21] [22] [23] [24]. Ces particules contiennent toutes l’apoB100, en tant qu’apoprotéine majeure, et du cholestérol, en proportion différente ; de plus, elles favorisent le processus athéroscléreux au cours de toutes les étapes clés : initiation, progression et rupture [2] [10] [17] [20-24] [25] [26].
Suite à leur pénétration et leur rétention préférentielles, ces lipoprotéines athérogènes s’accumulent dans l’espace sous-endothélial, déclenchant le recrutement et l’infiltration de monocytes circulants dans l’intima, conduisant à la constitution de stries graisseuses, dites stries lipidiques, à la surface luminale [17] [23-26] [27] [28].
Dans les stries lipidiques déjà visibles chez des fœtus de mères hypercholestérolémiques, la présence de macrophages est toujours associée à celle de LDL oxydées, alors qu’il n’y a pas de macrophages dans les lésions riches en LDL natives, non oxydées [29].
Il est ainsi possible d’établir la chronologie des premiers événements de l’athérosclérose :
- infiltration et rétention des lipoprotéines athérogènes ;
- modifications oxydatives des LDL ;
- recrutement monocytaire (fig. 12-1).
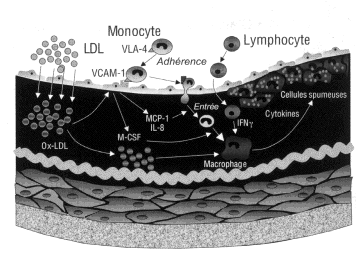
Fig. 12-1.-Étapes clés de la formation des stries lipidiques, précurseurs des plaques d’athérome
L’accumulation de LDL-cholestérol dans l’intima artérielle est la conséquence de l’augmentation de la perméabilité endothéliale. Les LDL retenues par la matrice extracellulaire sont soumises à des altérations oxydatives, avec formation de nombreux lipides oxydés pro-inflammatoires et modifications covalentes de l’apoB. Simultanément les cellules endothéliales activées expriment des protéines d’adhésion qui se lient aux monocytes : ceux-ci traversent l’endothélium et se transforment en macrophages dans l’intima. A leur tour les macrophages expriment divers récepteurs qui reconnaissent les LDL oxydées (ou récepteurs scavenger) par leur charge électronégative, facilitant ainsi leur internalisation avec accumulation intracellulaire de cholestérol et formation de cellules spumeuses. Ces cellules produisent des facteurs pro-inflammatoires et pro-thrombogénes qui favorisent la progression de la plaque et la fragilisation de sa matrice extracellulaire.
L’une des fonctions essentielles de l’endothélium capillaire normal est de faciliter le transport des protéines, lipoprotéines et autres composantes solubles de masse moléculaire élevée de la lumière du vaisseau vers les tissus sous-jacents. Les vésicules de pinocytose appelées cavéoles participent à ce transfert par le processus de transcytose [25, 26]. La pénétration et la rétention sous-endothéliale des lipoprotéines contenant l’apoB100 constituent les événements initiateurs de l’athérogenèse [26, 28].
La rétention spécifique de ces particules à des sites de prédilection résulte de l’interaction électrostatique entre les protéoglycanes de la matrice extracellulaire et les régions basiques de l’apoB100 (notamment les résidus 3 359 à 3 369) [27]. Cette interaction peut être également favorisée par la lipoprotéine lipase qui exerce une fonction de bridging entre les particules LDL et protéoglycanes [27,28].
L’accumulation des LDL au niveau intimal reflète un déséquilibre entre leurs flux d’entrée et de sortie [8, 23, 27]. Il peut résulter d’une augmentation de la perméabilité endothéliale, d’une diminution de celle de la média, de la présence de protéoglycanes ou de collagène qui fixent les LDL, de la dégradation irréversible des LDL ou d’une combinaison de ces processus [28]. Une concentration plasmatique élevée de LDL, comme celle observée dans les hyperlipidémies de phénotype IIA (hypercholestérolémie) et IIB (hypercholestérolémie et hypertriglycéridémie), favorise également un flux d’entrée élevé de ces particules [22-24, 26]. Enfin, les facteurs hémodynamiques (pression, forces de cisaillement à la paroi, turbulences, stagnation d’écoulement) influent sur le transfert des LDL à travers la paroi et sur le temps de résidence des particules athérogènes [23, 26] [30].
Normalement, à la surface de l’endothélium vasculaire, site commun d’action des facteurs de risque cardiovasculaire, les forces de cisaillement laminaires constantes dues au flux sanguin maintiennent les cellules endothéliales dans un phénotype anti-inflammatoire, anti-oxydant et antithrombotique. En revanche, l’activation inflammatoire de l’endothélium se produit de façon caractéristique au niveau des sites de prédilection dans l’arbre artériel et en particulier au niveau des bifurcations artérielles, des courbures et de l’origine des branches où la surface endothéliale est soumise à des forces de cisaillement faibles ou négatives [30] ; ces forces peuvent varier considérablement au cours du cycle cardiaque.
Des forces de cisaillement négatives peuvent directement induire une activation endothéliale qui a trois caractéristiques principales :
- une perméabilité endothéliale aux solutés significativement augmentée facilitant la pénétration des lipoprotéines athérogènes ;
- une expression d’intégrines ou protéines d’adhésion (telles que VCAM-1 et ICAM-1) à la surface de l’endothélium, participant à la réaction inflammatoire.
En effet, ces protéines se lient spécifiquement aux récepteurs des cellules inflammatoires comme les monocytes tels que les intégrines VLA-4 (a4ß2) et LFA-1 (aLß2, CD IIa/CD18) permettant ainsi leur adhésion puis leur pénétration dans l’intima artérielle ;
- l’induction d’un stress oxydatif qui altère de nombreuses fonctions de l’endothélium dont le tonus vasomoteur [30]. Ce phénomène biologique complexe est également induit par les effets de divers facteurs de risque (hypertension, hypercholestérolémie, diabète et tabac), via une réduction de la biodisponibilité du monoxyde d’azote.Une réduction de la biodisponibilité du NO peut résulter de l’augmentation de la dégradation du NO par les radicaux libres (appelés ROS). Le radical libre le plus impliqué dans l’inactivation du NO est l’anion superoxyde (O2•-) qui semble être produit principalement par la NADH/NADPH oxydase endothéliale. L’activité de cet enzyme est régulée par les forces mécaniques (ou forces de cisaillement), par les cytokines et des hormones ; tous ces stimuli entraînent une augmentation soutenue de l’activité de l’oxydase. Les radicaux libres tels que O2•- peuvent réagir avec le NO pour former le peroxynitrite, agent qui catalyse l’oxydation des acides gras polyinsaturés, composants essentiels des phospholipides et des esters de cholestérol dans les particules LDL.
L’activation endothéliale et le stress oxydant avec lequel il est intimement associé constituent donc des facteurs clés dans la formation intimale des lésions contenant des lipides oxydés [17,23, 26, 30].
MODIFICATION DES LDL ET FORMATION DES CELLULES SPUMEUSES
LE RÉCEPTEUR AUX LDL
Pour se transformer en cellules spumeuses, les macrophages infiltrés dans le sous-endothélium doivent capter et internaliser de grandes quantités de LDL.
Or, très tôt, Brown et Goldstein [31] ont découvert que le récepteur cellulaire des LDL, dont le ligand est l’apolipoprotéine-B (apoB) pour les LDL et l’apoE pour les VLDL, est soumis à une régulation négative (down-regulation) : lorsque la concentration en cholestérol intracellulaire augmente au-delà d’un certain seuil, la synthèse des récepteurs cesse et la captation et l’internalisation des LDL décroissent. Cette régulation métabolique maintient les taux intracellulaires d’esters de cholestérol très inférieurs à ceux retrouvés dans les cellules spumeuses. La voie de captation des LDL par le récepteur des LDL ne peut donc pas expliquer la transformation des macrophages ou des cellules musculaires lisses en cellules spumeuses.
Brown et Goldstein ont démontré que les LDL devaient subir des modifications entraînant un changement de conformation de l’apoB, la perte de reconnaissance par le récepteur des LDL et leur captation par d’autres récepteurs [32].
RÉCEPTEURS SCAVENGER
Pour se transformer en cellules spumeuses, les macrophages internalisent de grandes quantités de LDL oxydées par l’intermédiaire de récepteurs dits éboueurs (scavenger) (SR-AI, SR-AII, CD36, CD68) qui, à l’inverse du récepteur des LDL normales, ne sont pas sous le contrôle négatif du contenu intracellulaire en cholestérol [33].
L’importance de ces récepteurs dans l’athérosclérose a été soulignée par des expériences chez les souris apoE-/- déficientes pour les récepteurs SRA [34] ou CD36 [35] qui développent nettement moins de lésions athéroscléreuses que les souris apoE-/- dotées de récepteurs normaux.
Le récepteur scavenger de type A a été cloné [36]. Il en existe deux types, I et II. Le récepteur de type I se présente sous la forme d’un trimère ayant l’aspect d’une tige hérissée à l’extérieur de la cellule. Cette tige comprend trois domaines successifs : une hélice a, une séquence proche du collagène qui lie les LDL oxydées, puis un domaine C-terminal riche en cystéine (qui est tronqué sur le récepteur de type II). Récemment, un nouveau récepteur dénommé LOX-1, appartenant à la famille des lectines de type C, et capable de reconnaître et d’internaliser les LDL oxydées, a été identifié sur les cellules endothéliales [37].
AUTRES MODES DE CAPTATION DES LDL
D’autres modes de captation des LDL par le macrophage conduisent à une surcharge intracellulaire en esters de cholestérol : il s’agit de la phagocytose et des complexes immuns.
Phagocytose
Les LDL présentent une affinité importante pour les glycosaminoglycanes de la matrice extracellulaire, avec lesquels elles forment des complexes insolubles que le macrophage est capable de phagocyter [38]. On observe aussi la formation d’agrégats de LDL dont la phagocytose est déclenchée par la liaison de plusieurs particules LDL à leurs récepteurs spécifiques.
Complexes immuns
Desauto-anticorps dirigés contre les résidus lysine conjuguésà des fragments d’acide gras, tels que le malondialdéhyde, produits in vivo au cours des processus oxydatifs, ont été retrouvésdans le sang d’animaux normaux et hypercholestérolémiques, ainsi que chez des sujets sainsou coronariens [39]. Cesauto-anticorpspourraient reconnaître des LDL faiblement oxydées et former un complexe immun, qui serait alors capté avec une forte affinité par le récepteur du fragment Fc des immunoglobulines, présent sur les macrophages.
OXYDATION DES LDL AU NIVEAU INTIMAL
L’oxydation des composantes lipidiques et protéiques des LDL est une étape essentielle du processus athéroscléreux. Elle se produit majoritairement in situ, dans la paroi [40]. On ne retrouve en effet que de très faibles quantités de LDL oxydées circulantes alors qu’elles sont présentes en abondance dans la plaque athéroscléreuse [41].
L’oxydation des LDL peut être provoquée chimiquement in vitro (par exemple par incubation de LDL natives en présence de malondialdéhyde, de radicaux libres ou d’extraits de fumée de cigarette). Les LDL peuvent être oxydées au contact des cellules endothéliales, des CML ou des macrophages [42]. Schématiquement (fig. 12-2), les étapes de l’oxydation lipidique des LDL comportent [43] :
- le démarrage du mécanisme par un radical libre oxygéné (ROS) qui se traduit par une peroxydation lipidique à la surface de la LDL. Cette peroxydation est au départ limitée et peut être induite par tous les facteurs cités précédemment ;
- la propagation du phénomène dépend en partie de l’enzyme PAF-AH (Platelet Activating Factor-Acetylhydrolase ou lipoprotein-associated phospholipase A2) [44]. Cette enzyme, principalement associée aux LDL, possède une activité phospholipase A2. Après peroxydation d’une double liaison (C=C) au sein d’un acide gras polyinsaturé, composant d’une moléculed’ester de cholestérol, dephospholipide ou de triglycéride, cette activité peut provoquer une amplification de la peroxydation lipidique, conduisant à une fragmentation d’acides gras polyinsaturés avec génération d’aldéhydes et de cétones. Les cétones sont éliminées, mais les aldéhydes se lient aux résidus lysine de l’apoB100, ligand clé du récepteur cellulaire des LDL. Il s’ensuit une modification covalente de l’apoB100 avec augmentation de sa charge négative, entraînant une perte de la reconnaissance par le récepteur des LDL natives, mais lui conférant la capacité de se lier au récepteur scavenger [10,24, 39-43].
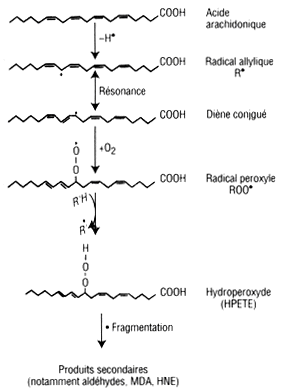
Fig. 12-2 LDL retenues au niveau intimal subissant une modification oxydative en conséquence de leur interaction avec les espèces réactives de l’oxygène
À titre d’exemple, l’acide arachidonique, composant polyinsaturé des phospholipides et des esters de cholestérol des LDL, est transformé en hydroperoxyde lipidique, l’acide hydroperoxy-eicosatétraénoïque (HPETE), suite à sa perte d’un atome d’hydrogène lors d’une attaque radicalaire, et de l’addition de l’oxygène moléculaire. La fragmentation de cet hydroperoxyde peut ensuite donner lieu à la formation de produits secondaires tels que des aldéhydes à courte chaîne possédant des activités chimiques et biologiques élevées. La fragmentation des hydroperoxydes lipidiques est favorisée par le stress oxydant, incluant les cations métalliques tels que le cuivre (Cu2+) et le fer (Fe3+), et des activités enzymatiques (PAF-AH).
L’oxydation des LDL par les cellules endothéliales, mais pas par les CML ou par les macrophages, nécessite le contact entre LDL et cellules. La production cellulaire de radicaux libres pourrait être à l’origine de l’oxydation des LDL [42]. La 15-lipoxygénase semble y jouer un rôle important [45] : dans la plaque d’athérome, il existe en effet une colocalisation des LDL oxydées et des ARNm codant pour cette enzyme.
La modification biologique des LDL par les différents types cellulaires de la plaque peut expliquer la présence de LDL oxydées, révélée par immunomarquage [10]. Il est également possible que les LDL finissent par s’oxyder lorsque leur temps de séjour dans l’intima est augmenté, la pO2 dans le sous-endothélium étant relativement élevée (sensiblement égale à celle du sang). De plus, des travaux récents rapportent que les LDL immobilisées dans la matrice extracellulaire sous-endothéliale peuvent subir une attaque enzymatique, avec génération de particules non oxydées, de 10 à 200 nm de diamètre, capables d’activer le complément et d’induire une réponse inflammatoire au niveau des cellules vasculaires et des macrophages [26] [46].
Rôle des LDL oxydées dans l’inflammation et l’athérosclérose
Les études expérimentales ont permis d’établir de façon claire un lien entre hypercholestérolémie et présence d’une réaction inflammatoire dans le tissu vasculaire [24], [47]. De nombreuses études ont également été réalisées afin d’évaluer l’effet des LDL oxydées comme agent inflammatoire. Celles-ci ont un pouvoir chimioattractant sur les monocytes, favorisent leur différenciation en macrophages résidents, et, en revanche, inhibent la motilité de ces derniers [10, 24].
L’exposition à des LDL oxydées de cellules mononucléées du sang humain, constituées de monocytes, de lymphocytes T et occasionnellement de lymphocytes B, se traduit par l’activation élective de la population lymphocytaire T, attestée par l’expression accrue de récepteurs de l’IL-2 et des antigènes HLA-DR sur les cellules T [48]. L’administration in vivo chez la souris de LDL légèrement oxydées, dites minimally modified LDL (MM-LDL) provoque l’induction rapide de Monocyte-Colony Simulating Factor (M-CSF) dans le sang et des gènes pro-inflammatoires codant pour JE (l’homologue murin du facteur chimiotactique des monocytes, MCP-1) et d’autres protéines de l’inflammation dans les tissus.
Cette réponse est identique à celle induite par un régime athérogène et renforce l’idée que les produits d’oxydation dérivés des lipides sont à l’origine de la réaction inflammatoire [49]. Mais cette réponse pourrait être contrôlée génétiquement, car elle n’est pas retrouvée chez les souris de la souche C3H qui ne développent pas de lésions athéromateuses même lorsqu’elles sont soumises à un régime hypercholestérolémiant [50] [51].
Les LDL oxydées pourraient engendrer des réactions auto-immunes, comme en témoigne la présence chez l’homme d’anticorps circulants spécifiques dirigés contre ces molécules immunes [52]. L’étude de Salonen et coll. [53] rapporte des taux d’auto-anticorps anti-LDL oxydées significativement plus élevés chez des patients souffrant d’athérosclérose carotidienne évolutive par rapport à un groupe contrôle du même âge. Le titre sérique en anticorps anti-LDL oxydées (anticorps anti-malondialdéhyde) est un marqueur prédictif indépendant de la progression de la maladie athéromateuse.
Des travaux récents montrent que ces auto-anticorps reconnaissent également des épitopes spécifiques de l’oxydation présents sur les cellules apoptotiques, qui contribuent très certainement à l’homéostasie physiologique des cellules mortes par apoptose ou par nécrose, après avoir subi des modifications oxydatives [54]. Il s’agit d’anticorps naturels présents dans l’organisme dès les premiers instants de la vie qui seraient sélectionnés en réponse à une charge excessive en antigènes spécifiques de l’oxydation, comme cela se passe au cours de l’athérosclérose.
Apoptose et athérosclérose
Les plaques d’athérosclérose contiennent de nombreux débris cellulaires provenant de la nécrose des macrophages mais elle est aussi le siège d’intenses processus apoptotiques [55]. L’apoptose survient dans tous les types cellulaires : ce sont essentiellement les macrophages et les lymphocytes T, mais les cellules endothéliales situées en aval de la sténose maximale et exposées à des faibles taux de cisaillement peuvent aussi être affectées [56]. La réaction inflammatoire détermine, en grande partie, le taux de cellules apoptotiques.
Les cytokines pro-inflammatoires peuvent induire ce processus par la production excessive de monoxyde d’azote conduisant à la formation de peroxynitrite [57]. L’expression de la NO synthase inductible est directement corrélée à la survenue du phénomène dans la plaque humaine [58]. A l’inverse, l’expression locale de cytokines anti-inflammatoires comme l’interleukine-10 est associée à une diminution de l’expression de la NO synthase inductible et à une diminution de l’apoptose [58].
Les données les plus actuelles font jouer un rôle déterminant à l’apoptose dans la formation du thrombus à l’origine des syndromes coronariens aigus [55].
Celle des macrophages est fréquemment observée en bordure du noyau lipidique acellulaire, ce qui suggère que la mort des macrophages par ce processus contribue à la croissance du noyau lipidique dans lequel s’accumulent des microparticules apoptotiques, fragments membranaires de cellules mortes.
Le rôle fonctionnel majeur de l’apoptose dans la plaque d’athérome est lié au potentiel procoagulant des cellules et microparticules apoptotiques.
Remodelage vasculaire et formation de la chape fibro-musculaire
Les manifestations cliniques graves de la maladie athéromateuse (mort subite, infarctus myocardique ou cérébral, angor instable) ont peu à voir avec la taille de la plaque, mais sont essentiellement dues à son instabilité. Celle-ci se caractérise, au moment de l’incident, par la survenue d’un thrombus luminal au contact d’une rupture de la chape fibreuse (60 % des cas) ou d’une « érosion » endothéliale (40 % des cas) [59] [60]. La plaque se développe longtemps sans altérer le calibre vasculaire, ce qui remet en question l’intérêt diagnostique de l’angiographie conventionnelle [61]. Ce processus d’adaptation des vaisseaux athéroscléreux a été dénommé remodelage vasculaire ou élargissement compensateur. Toutefois, lorsque la masse intimale excède 40 % de la surface totale de la paroi, le remodelage excentrique de l’artère n’est plus suffisant pour contenir la plaque : son développement se fait alors aux dépens de la lumière artérielle et conduit à son obstruction progressive.
L’une des caractéristiques essentielles des plaques stables est la présence d’une chape fibreuse épaisse enrichie en CML et en collagène [60]. Les CML de la chape proviennent de cellules ayant migré à partir de la média à travers la limitante élastique interne et proliféré dans l’intima.
La structure de la chape fibro-musculaire constituée, souvent organisée en unités lamellaires avec des CML redevenues contractiles, laisse penser qu’il s’agit là d’un phénomène de type cicatriciel, la média originelle étant fréquemment atrophiée ou inexistante, suite probablement à l’activité des métalloprotéases sécrétées par les macrophages.
Les cellules adultes quiescentes de la média présentent un phénotype contractile, leur taux de renouvellement est très bas et la sécrétion des protéines de la matrice extracellulaire interrompue. En revanche, après avoir migré dans l’intima, elles adoptent un phénotype sécrétoire : elles prolifèrent, synthétisent et sécrètent en abondance les protéines extracellulaires. Toutefois, les CML provenant de la chape fibreuse de plaques athéroscléreuses sont beaucoup plus sensibles à l’apoptose que les CML issues de la média [62], ce qui pourrait expliquer l’instabilité de certaines plaques.
Le facteur stimulant la prolifération des cellules musculaires lisses est probablement le PDGF. Les cellules endothéliales, les macrophages et les CML de la plaque en sécrètent. Mais il existe d’autres facteurs de croissance candidats, en particulier le bFGF et l’IGF-1. L’IGF-1 est un puissant facteur de survie des CML. Une diminution du nombre de récepteurs de l’IGF-1 à la surface des CML explique leur plus forte propension à l’apoptose comparée aux CML provenant d’une média normale [63].
Deux mécanismes d’action ont donc été suggérés pour expliquer l’effet des facteurs de croissance :
- une action paracrine : après libération par les cellules endothéliales et macrophages environnants, le PDGF agit sur les CML ;
- une action autocrine : la cellule sécrète elle-même l’agent qui agit sur sa propre croissance.
Conclusion
La fin du XXe siècle a révélé une évolution rapide des concepts physiopathologiques de l’athérosclérose.
Le caractère inflammatoire indéniable de la maladie oriente vers des stratégies thérapeutiques spécifiques au cours des accidents coronariens et cérébraux.
La réduction thérapeutique des lipoprotéines athérogènes par un traitement hypolipémiant représente un élément essentiel de cette stratégie, limitant la disponibilité du substrat pour l’oxydation et la formation de lipides oxydés pro-inflammatoires.
Par contre, l’utilisation de molécules antioxydantes visant à protéger les lipides insaturés de l’oxydation s’est pour le moment soldée par un échec [64].
La reconnaissance du rôle stabilisateur des CML dans la plaque a conduit à abandonner l’idée d’agir sur la plaque en inhibant la prolifération de ces cellules.
La capacité de la plaque à croître en remodelant l’artère, sans créer de sténose luminale, amène à développer des techniques d’imagerie (ultrasons, imagerie par résonance magnétique) capables de visualiser la paroi et d’en identifier les composants. Enfin, la découverte de l’apoptose comme déterminant majeur de la thrombose laisse entrevoir de nouvelles possibilités pour prévenir la survenue d’accidents ischémiques aigus.
Liste des abréviations
Monocyte-Colony Simulating Factor
Interleukine
minimally modified
Monocyte Chemoattractant Protein
Monoxyde d’azote
Platelet Derived Growth Factor
M-CSF
IL
MM-LDL
MCP
NO
PDGF
Article extrait de l’ouvrage « L’athérosclérose Physiologie, diagnostics, thérapeutiques J.-F. Toussaint, M.-P. Jacob, L. Lagrost, J. Chapman, sous l’égide de la Société Française d’Athérosclérose », © Masson, Paris, 2003
[1] Ford ES., Giles W.H., Dietz W.H.- Prevalence of the Metabolic Syndrome among US adults. Findings from the Third National Health and Nutrition Examination Survey. JAMA, 2002 287 : 356-359.
[2] Libby R- Current concepts of the pathogenesis of the acute coronary syndromes. Circulation, 2001, 104 365-372.
[3] Ruffer MA.- On arterial lesions found in Egyptian mummies. J Pathol Bacteriol, 1911 :15 : 453.
[4] Goldman D.S.- Cholesterol revisited. Molecule, medicine and media. Arteriosclerosis, 1989 : 9 : 430-438.
[5] Fye W.B.- A historical perspective on atherosclerosis and coronary artery disease. In : Fuster V., Ross R., Topol E.J. Atherosclerosis and Coronarv Artery Disease. Lippincott-Raven, Philadelphia, 1996, 1-12.
[6] Stary H.C., Chandler A.B., Dinsmore RE. et coll. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis : a report from the committee on vascular lesions of the Council on Arteriosclerosis, Amencan Heart Association. Circulation. 1995 : 92 : 1355-1374.
[7] Schwenke DC., Carew TE.- Initiation of atherosclerotic lesions in cholesterol-fed rabbits, I : focal increases in arterial LDL concentration precede development of fatty streak lesions. Arreriosclerosis, 1989 9 : 895-907.
[8] Schwenke DC., Carew TE.- Initiation of atherosclerotic lesions in cholesterol-fed rabbits, Il : Selective retention of LDL vs. selective increases in LDL permeability in susceptible sites of arteries. Arteriosclerosis, 1989 : 9 : 908-918.
[9] Jonasson L., Holm J., Skalli O., Bondjers G., Hans-son G.- Regional accumulations of T cells, macrophages, and smooth muscle cells in the human atherosclerotic plaque. Atherosclerosis, 1986 : 6 : 131-138.
[10] Steinberg D., Parthasarathy S., Carew TE., Khoo J.C., Witztum J.L.- Beyond cholesterol. Modifications of low-density lipoprotein that increase its atherogenicity. N Engl J Med, 1989 : 320 : 915-924.
[11] Hansson G.K., Holm J., Jonasson L.- Detection of activated T lymphocytes in the human atherosclerotic plaque. Am J Pathol, 1989 135 :169-175.
[12] Virchow R.- Phlogose und Thrombose im Gefassystem. In : Gesammelte Abhanndlungen zür wissensehaftlichen Medizin. F Meidinger Sohn and Company, Frankfurt-am-Main, 1856 :458-521.
[13] Ross R., Glomset JA.- The pathogenesis of atherosclerosis. N Engl J Med, 1976 :295 :420-425.
[14] Taylor KE.. Glagov S., Zarins C.K.- Preservation and structural adaptation of endothelium over experimental foam cell lesions. Quantitative ultrastructural study. Arteriosclerosis, 1989 9 : 881-894.
[15] Weissberg P.L.. Clesham G.J., Bennett M.R.- Is vascular smooth muscle cell proliferation beneficial ? Lancet, 1996 347 : 305-307.
[16] Davies M.J.- Stability and instability : Two faces of coronary atherosclerosis – The Paul Dudley White Lecture 1995. Circulation, 1996 :94 : 2013-2020.
[17] Ross R.- Atherosclerosis-an inflammatory disease. N Engl J Med, 1999:340:115-126.
[18] Shi W., Haberland ME.. Jien ML.. Shih DM., Lusis A.J.- Endothelial responses to oxidized lipoproteins determine genetic susceptibility to atherosclerosis in mice. Circulation, 2000 :102 :75-81.
[19] Steinberg D., Gotto A.M. Jr. Preventing coronary artery disease by lowering cholesterol levels : fifty years from bench to bedside. JAMA. 1999 : 282:2043-2050.
[20] Genest J. Jr., Cohn J.S.- Epidemiological evidence linking plasma lipoprotein disorders to atherosclerosis and other diseases. In : Barter PJ., Rye KA. Plasma Lipids and their role in disease. Taylor and Francis, London, 1999 : 46-48.
[21] Hodis N.N., Mack W.J.- Triglyceride-rich lipoproteins and progression of atherosclerosis. Eur Heart J, 1998 :19 (suppl. A) :40-44.
[22] Chapman M.J.- Atherogenicity of Low Density Lipoprotein : mechanisms. In : Barter PJ., Rye KA. Plasma Lipids and their role in disease. Taylor and Francis, London. 1999.
[23] Nordestgaard B.G., Nielson LB.- Atherosclerosis and arterial influx of lipoproteins. Curr Opin Lipidol, 1994 :5 : 252-257.
[24] Young S.G., Parthasarathy S.- Why are low-density lipoproteins atherogenic ? West J Med, 1994, 160 : 153-164.
[25] Vasile E., Simionescu M., Simionescu N.- Visualization of the binding, endocytosis, and transcytosis of low-density lipoprotein in the arterial endothelium in situ. J Cell Biol, 1983 : 96:1677-1689.
[26] Williams K.J., Tabas I.- The response-to-retention hypothesis of early atherogenesis. Arterioscler Thromb Vasc Biol, 1995 :15 :551-561.
[27] Carew TE., Pittman R.C., Marchand ER., Steinberg D.- Measurement in vivo of irreversible degradation of low density hipoprotein in the rabbit aorta : predominance of intimal degradation. Arteriosclerosis, 1984 :4 : 214-224.
[28] Boren J., Gustafsson M., Skalen K., Flood C., Inneranity T.L.- Role of extracellular retention of low density lipoproteins in atherosclerosis. Curr Opin Lipidol, 2000:11:451-456.
[29] Napohi C., D’Armiento EP., Mancini EP. et coll.- Fatty streak formation occurs in human fetal aortas and is greatly enhanced by maternal hypercholesterolemia. Intimal accumulation of low density lipoprotein and its oxidation precede monocyte recruitment into early atherosclerotic lesions. J Clin Invest, 1997 :100 : 2680-2690.
[30] Gimbrone MA. Jr.- Endothelial dysfunction, hemodynamic forces, and atherosclerosis. Thromb Haemost, 1999 :82 : 722-726.
[31] Brown MS., Goldstein J.L.- A receptor-mediated pathway for cholesterol homeostasis. Science, 1986 232 :34-47.
[32] Brown MS., Goldstein J.L., Krieger M., Ho Y.K., Anderson R.G.- Reversible accumulation of cholesteryl esters in macrophages incubated with acetylated lipoproteins. J Cell Biol, 1979 82 : 597-613.
[33] de Winther M.P., van Dijk KW., Havekes L.M., Hofker M.H.- Macrophage scavenger receptor class A : A multifunctional receptor in atherosclerosis. Arterioscler Thromb Vasc Biol, 2000 ; 20:290-297.
[34] Suzuki H.. Kurihara Y.. Takeya M. et coll.- A role for macrophage scavenger receptors in atherosclerosis and susceptibility to infection. Nature. 1997 : 386 : 292-296.
[35] Febbraio M., Podrez E.A., Smith J.D. et coll.- Targeted disruption of the class B scavenger receptor CD36 protects against atherosclerotic lesion development in mice. J Clin Invest, 2000:105:1049-1056.
[36] Kodama T., Freeman M., Rohrer L., Zabrecky J., Matsudaira P., Krieger M.- Type I macrophage scavenger receptor contains alpha-helical and collagen-like coiled coils. Nature, 1990 343 :531-535.
[37] Sawamura T., Kume N., Aoyama T. et coll.- An endothelial receptor for oxidized low-density lipoprotein. Nature, 1997 :386 : 73-77.
[38] Hurt E., Bondjers G., Camejo G.- Interaction of LDL with human arterial proteoglycans stimulates its uptake by human monocyte-derived macrophages. J Lipid Res, 1990 :31 : 443-454.
[39] PalinskiW.,Rosenfeld ME., Yla-Herttuala S. et coll.- Low density lipoprotein undergoes oxidative modification in vivo. Proc Natl Acad Sci USA, 1989 86 :1372-1376.
[40] Steinberg D.- Oxidative modification of LDL and atherogenesis. Circulation, 1997 95 :1062-1071.
[41] Chisolm GM., Steinberg D.- The oxidative modification hypothesis of atherogenesis : an overview. Free Radic Biol Med, 2000 :28:1815-1826.
[42] Chisolm GM. 3rd, Hazen S.L., Fox P.L., Cathcart M.K.- The oxidation of lipoproteins by monocytes-macrophages. Biochemical and biological mechanisms. J Biol Chem, 1999 ; 274 : 25959-25962.
[43] Witztum J.L., Steinberg D.- Role of oxidized low density lipoprotein in atherogenesis. J Clin Invest, 1991 :88:1785-1792.
[44] Navab M., Berliner JA., Watson AD. et coll.- The Yin and Yang of oxidation in the development of the fatty streak. Arterioscler Thromb Vasc Biol, 1996,16 :831-842.
[45] Yla-Herttuala S., Rosenfeld ME., Parthasarathy S. et coll.- Colocalization of 15-lipoxygenase mRNA and protein with epitopes of oxidized low density lipoprotein in macrophage-rich areas of atherosclerotie lesions. Proc Natl Acad Sci USA 1990 :87 : 6959-6963.
[46] Klouche M., Rose-John S., Schmiedt W., Bhakdi S.- Enzymatically degraded. nonoxidized LDL induces human vascular smooth muscle cell activation, foam cell transformation, and proliferation. Circulation, 2000 101:1799-1805.
[47] Liao E, Andalibi A., Qiao J.H., Allayee H., Fogelman A.M., Lusis A.J.- Genetic evidence for a common pathway mediating oxidative stress, inflammatory gene induction, and aortic fatty streak formation in mice. J Clin Invest, 1994 : 94 : 877-884.
[48] Frostegaard J., Wu R., Giscombe R., Holm G., Lefvert A.K., Nilsson J.- Induction of T-cell activation by oxidized low density lipoprotein. Arterioscler Thromb, 1992 :12 : 461-467.
[49] Li H., Cybulsky MI., Gimbrone M.A.J., Libby P.- An atherogenic diet rapidly induces VCAM-1, a cytokine-regulatable mononuclear leukocyte adhesion molecule, in rabbit aortic endothelium. Arterioscler Thromb, 1993 :13:197-204.
[50] Liao F., Berliner JA., Mehrabian M. et coll.- Minimally modified low density lipoprotein is biologically active invivo in mice. J Clin Invest, 1991 ; 87:2253-2257.
[51] Berliner JA., Navab M., Fogelman A.M. et coll.- Atherosclerosis : basic mechanisms. Oxidation, inflammation, and genetics. Circulation, 1995 : 91:2488-2496.
[52] Palinski W., Witztum J.L.- Immune responses to oxidative neoepitopes on LDL and phospholipids modulate the development of atherosclerosis. J Intern Med, 2000 :247 : 371 -380.
[53] Salonen J.T., Yla-Herttuala S., Yamamoto R. et coll.- Autoantibody against oxidised LDL and progression of carotid atherosclerosis. Lancet, 1992, 339 : 883-887.
[54] Shaw P.X., Horkko S., Chang M.K. et coll.- Natural antibodies with the T15 idiotype may act in atherosclerosis, apoptotic clearance, and protective immunity. J Clin Invest, 2000 :105:1731-1740.
[55] Mallat Z., Tedgui A.- Current perspective on the role of apoptosis in atherothrombotic disease. Circ Res, 2001 :88 : 998-1003.
[56] Tricot O., Mallat Z., Heymes C., Lesèche G., Tedgui A.- Relation between endothelial cell apoptosis and blood flow direction in human atherosclerotic plaques. Circulation, 2000 : 101:2450-2453.
[57] Geng Y.J., Wu Q., Muszynski M., Hansson G.K., Libby P.- Apoptosis of vascular smooth muscle cells induced by in vitro stimulation with interferon-gamma, tumor necrosis factor-alpha, and interleukin- I beta. Arterioscler Thromb Vasc Biol, 1996 :16:19-27.
[58] Mallat Z., Heymes C., Ohan J., Faggin E., Lesèche G., Tedgui A.- Expression of interleukin-10 in human atherosclerotic plaques. Relation to inducible nitric oxide synthase expression and cell death. Arterioscler Thromb Vasc Biol, 1999 :19:611-616.
[59] Virmani R., Kolodgie ED., Burke A.P., Farb A.. Schwartz SM.- Lessons from sudden coronary death : a comprehensive morphological classification scheme for atherosclerotic lesions. Arterioscler Thromb Vasc Biol, 2000 :20:1262-1275.
[60] Davies M.J.- The composition of coronary- artery plaques. N Engl J Med, 1997:336:1312-1314.
[61] Glagov S., Weisenberg E., Zarins C.K., Stankunavicius R., Kolettis G.J.- Compensatory enlargement of human atherosclerotic coronary arteries. N Engl J Med, 1987 :316 :1371-1375.
[62] Bennett M.R., Evan GI., Schwartz SM.- Apoptosis of human vascular smooth muscle celîs derived from normal vessels and coronary atherosclerotic plaques. J Clin Invest, 1995 ; 95 : 2266-2274.
[63] Patel VA., Zhang Q.J., Siddle K. et coll.- Defect in insulin-like growth factor-1 survival mechanism in atherosclerotic plaque-derived vascular smooth muscle cells is mediated by reduced surface binding and signaling. Circ Res, 2001 :88 : 895-902.
[64] Neuzil J., Weber C., Kontush A.- The role of vitamin E in atherogenesis : Linking the chemical, biological and clinical aspects of the disease. Atherosclerosis, 2001 :157 : 257-283.